InVirtuoGen: a bit more insight into the model
Coming from physics, one of the most important insights I had was that the inductive biases of the architectures you use matter enormously. In physics, if you're modeling planetary orbits, you better have rotational symmetry built in. If you are working with particle clouds, your model should better be permutation invariant. The structure of your model should reflect the structure of your problem.
So when I arrived at In Virtuo Labs and saw their frontier model was based on GPT-style next-token prediction for molecules, something felt fundamentally wrong. They were generating molecules left-to-right, like writing a sentence. But molecules aren't sentences. There's no "first" atom in a benzene ring. The model was learning chemistry despite the architecture, not because of it.
The Search for Better Inductive Biases
Naturally, I started looking at what else was out there. The most promising thing I found was NVIDIA's GenMol - a masked discrete diffusion model. At least it wasn't pretending molecules had a left-to-right order. Instead, it would mask random tokens and gradually unmask them. Better, but still... once a token is unmasked during sampling, it is treated as fixed and no longer updated
Here's the thing: GenMol had no open code at the time for my to investigate. So I started building my own version. But instead of using their masked diffusion framework, I implemented it using discrete flow matching, mostly because I had just read the Gat et al. paper and thought it was elegant.
What started as "let me just recreate GenMol to understand it" turned into something unexpected.
My First Insight: Step Size Invariance
So one thing I realized quickly was that with masked diffusion models like GenMol, using more steps helps, but there is a hard limit: you cannot have more steps than you have tokens to unmask, unless you rely on some remasking heuristics, that did not seem to work well. So naturally I thought, that this was the time for my uniform source to shine. So I tested the performance of my discrete flow implementation as a function of the step size.
But what I discovered was depressing, with standard discrete flow sampling, changing the step size did... nothing. Whether I used 100 steps or 1000 steps, the model made the same total number of token changes. Look at this:
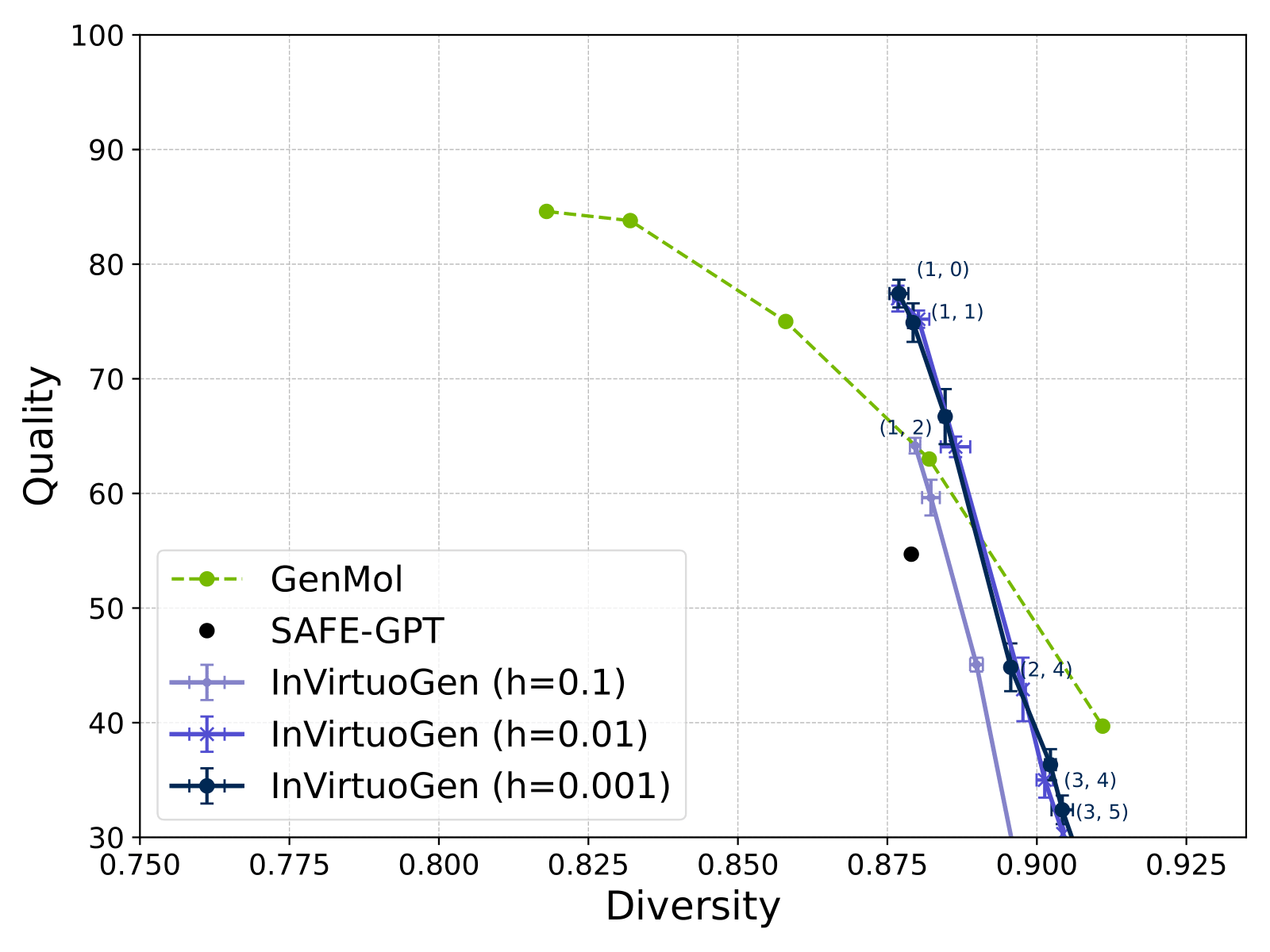
But then I had the idea to check how often the model actually changes a token. And what I saw was even more concerning: Whether you sample with granularity 0.01 or 0.001, the model changes the same number of tokens on average. This made little intuitive sense. If you take smaller steps, you should make more changes overall. But the model learned to compensate: smaller steps changed fewer tokens per step, keeping the total constant. It looked like the model was saying "I need exactly 50 edits to make this molecule, no more, no less."
This was depressing. If finer granularity doesn't help, then discrete flows have no advantage over masked diffusion. I almost gave up here.
The Sampling Hack That Saved Everything
Out of frustration (and honestly, a bit of desperation), I tried something theoretically wrong. Instead of using the proper discrete flow update equation:
Correct (but apparently useless) way:
$$ X_{t+h} \sim X_t + h\, v_\theta(X_t) $$What I tried in desperation:
$$ X_{t+h} \sim p_{1|T}(X_t) $$Basically, instead of taking small steps according to the velocity field, I just sampled fresh from the model's predicted distribution at each timestep. This shouldn't work better. It throws away the careful mathematical framework of flow matching.
But it did work better. Dramatically better. And suddenly, granularity mattered:
The dynamics completely changed. Instead of the model conserving its "edit budget," it now started with many simultaneous changes that gradually decreased as the molecule converged. It looked like actual refinement - messy at first, then progressively cleaner.
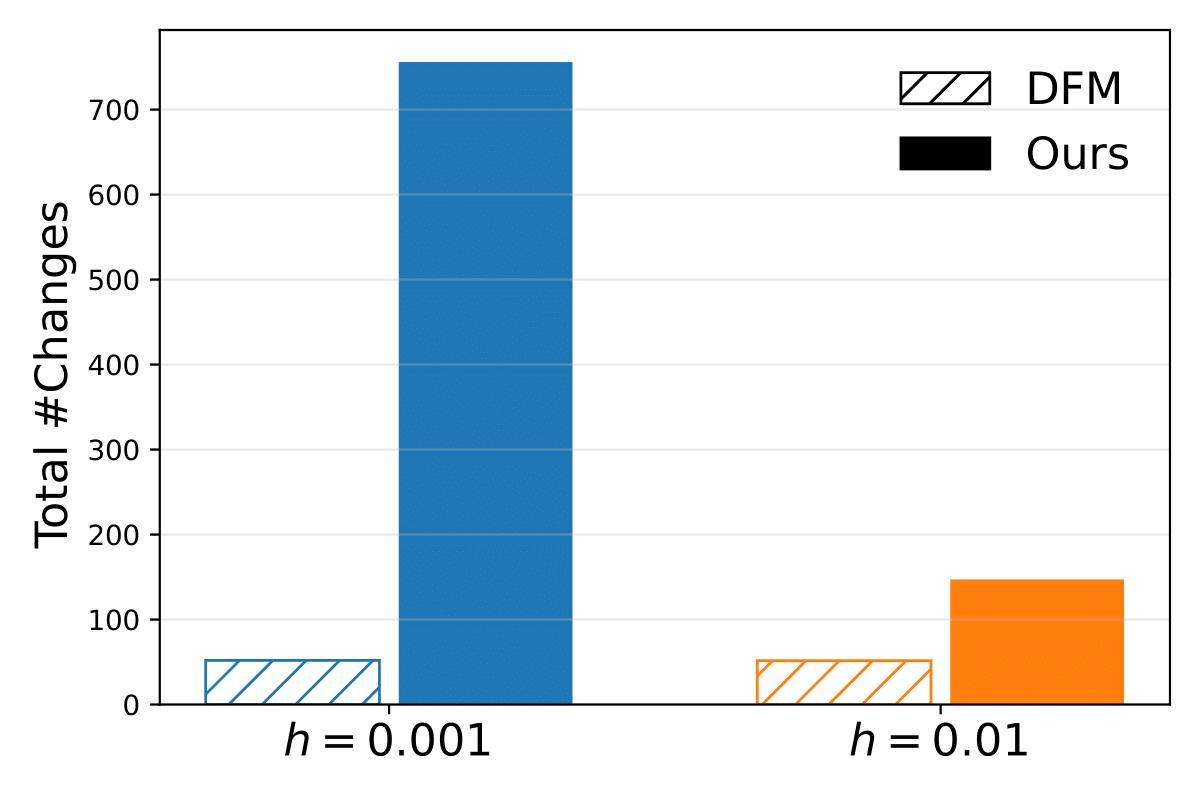
The standard discrete flow sampling expects the total number of jumps to be equal to the number of tokens, meaning that once a token is predicted, its value will likely not change anymore. However with our sampling method, in fact the number of jumps is significantly higher than the sequence length.
RL For Oracle Optimization: why GA plus PPO for flows
Established RL approaches in this area assume an autoregressive factorization: you can write down log p(x) as a sum of next token log probabilities and plug that into REINFORCE or PPO. That does not hold for discrete flows. There is no tractable log p(x) for the full sequence, and the policy acts on all positions at once, not left to right.
To make RL work, I adapted PPO to the flow setting by optimizing a time weighted objective on partially noised states. For each sequence, I sample timesteps t, construct a noised state \(x_t\) by interpolating between uniform tokens and the current sample, this then gives a MC estimate of the log probability of the sequence. Advantage comes from standardized oracle scores and then the standard PPO can be used (in practice actually BAPO is used for stability).
RL alone is not enough when oracle calls are scarce. I fuse a simple genetic algorithm that breeds high scoring molecules to create strong starting states \(x_{t=0}\). Crossover is done in fragmented SMILES space by swapping fragment blocks, then the flow refines the offspring. A small mutation budget on the best molecules maintains local exploration. An adaptive bandit biases sequence lengths that repeatedly yield high rewards while retaining exploration.
The PMO Benchmark
Beating GenMol on the PMO benchmark felt good, I won't lie. Especiallt given that I had no prior experience with reinforcment learning, and I believe I came up with a nice adaption of PPO to the discrete flow matching setting. But here's the thing - these benchmarks are weird. The "deco hop" task asks you to maximize the similarity to a specific molecule while decorating it. The "median molecules" tasks are essentially asking for mediocrity.
What actually matters is that the model can be directed toward specific objectives while maintaining chemical validity. The fact that we can do this with a single set of hyperparameters (while GenMol needs task-specific tuning) suggests we're doing something right. Or we got lucky. Probably both.
Limitations
- No stereochemistry - we completely ignore it, which is terrible for drug discovery
- The SA score we optimize for is a rough heuristic that often disagrees with actual chemists
- Our "drug-likeness" metrics are from 2012 - the field has moved on
- For fragment-constrained generation, we basically turn back into a masked model, negating our main advantage
- The model has no concept of 3D structure or protein binding - it's purely 1D
The real test will be whether actual drug discovery teams find this useful. I've shown it to a few medicinal chemists, and the reactions have been actually quite positive, and I was shocked that they did not rely on computational tools to do the heavy lifting.
Code and Reproducibility
Everything's on GitHub. There is a docker container for the code and significant effort was spent to make the results reproducible.
This is my first real project in drug discovery after transitioning from physics. I'm still learning, still making mistakes, and definitely still discovering how much I don't know. If you're working on similar problems or see flaws in my approach, I'd genuinely love to hear from you. Science is better when we admit what we don't know.
Find me at benno.kaech@icloud.com or the inevitable conference poster session where I'm explaining why our sampling hack works.